
How Rogue Jumping Genes Can Spur Alzheimer’s, ALS
Back in 2008, neurovirologist Renée Douville observed something weird in the brains of people who’d died of the movement disorder ALS: virus proteins.
But these people hadn’t caught any known virus.
Instead, ancient genes originally from viruses, and still lurking within these patients’ chromosomes, had awakened and started churning out viral proteins.
Our genomes are littered with scraps of long-lost viruses, the descendants of viral infections often from millions of years ago. Most of these once-foreign DNA bits are a type called retrotransposons; they make up more than 40 percent of the human genome.
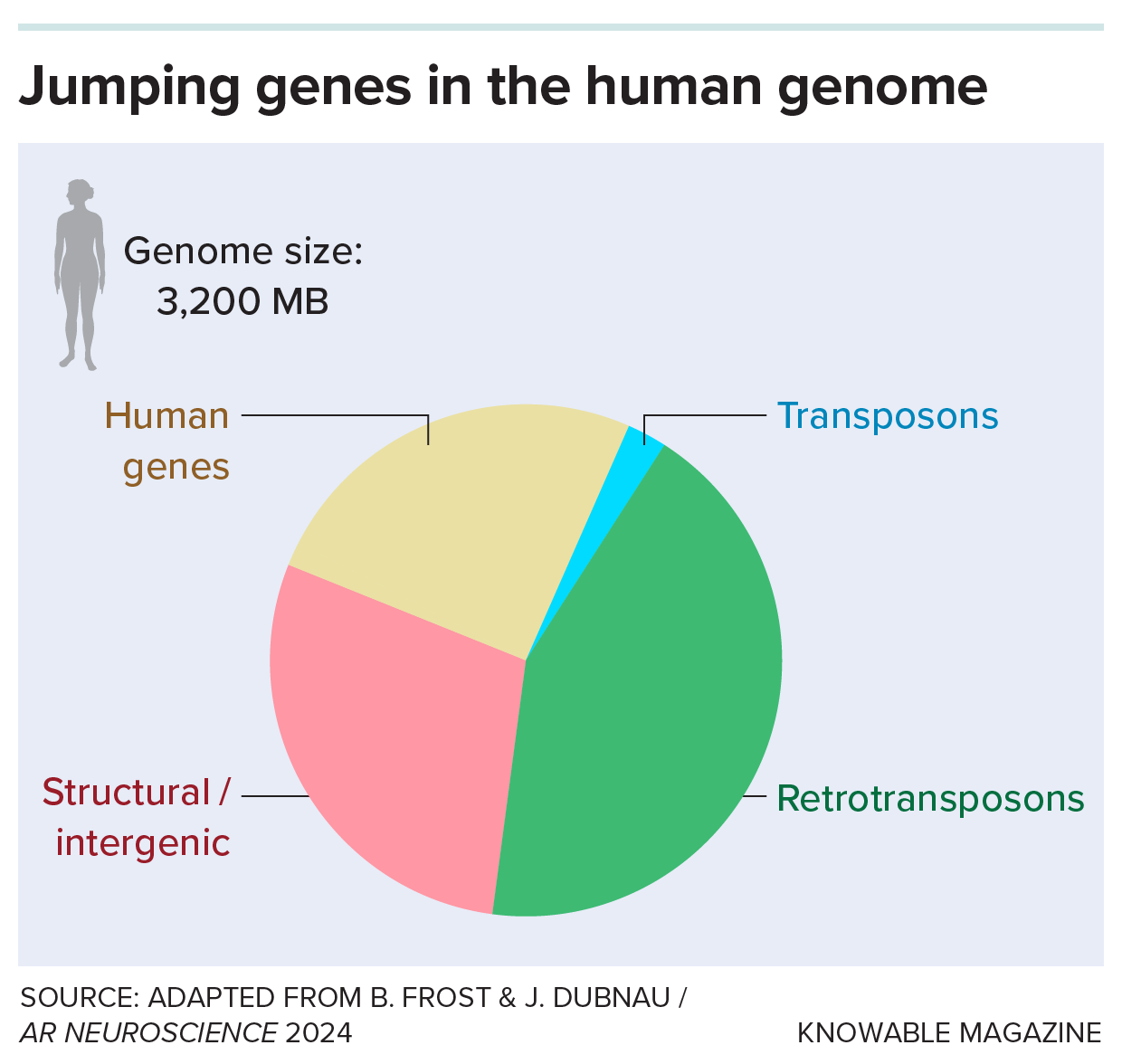
Many retrotransposons seem to be harmless, most of the time. But Douville and others are pursuing the possibility that some reawakened retrotransposons may do serious damage: They can degrade nerve cells and fire up inflammation and may underlie some instances of Alzheimer’s disease and ALS (amyotrophic lateral sclerosis, or Lou Gehrig’s disease).
The theory linking retrotransposons to neurodegenerative diseases — conditions in which nerve cells decline or die — is still developing; even its proponents, while optimistic, are cautious. “It’s not yet the consensus view,” says Josh Dubnau, a neurobiologist at the Renaissance School of Medicine at Stony Brook University in New York. And retrotransposons can’t explain all cases of neurodegeneration.
Yet evidence is building that they may underlie some cases. Now, after more than a decade of studying this possibility in human brain tissue, fruit flies and mice, researchers are putting their ideas to the ultimate test: clinical trials in people with ALS, Alzheimer’s and related conditions. These trials, which borrow antiretroviral medications from the HIV pharmacopeia, have yielded preliminary but promising results.
Meanwhile, scientists are still exploring how a viral reawakening becomes full-blown disease, a process that may be marked by what Dubnau and others call a “retrotransposon storm.”
Genes that jump
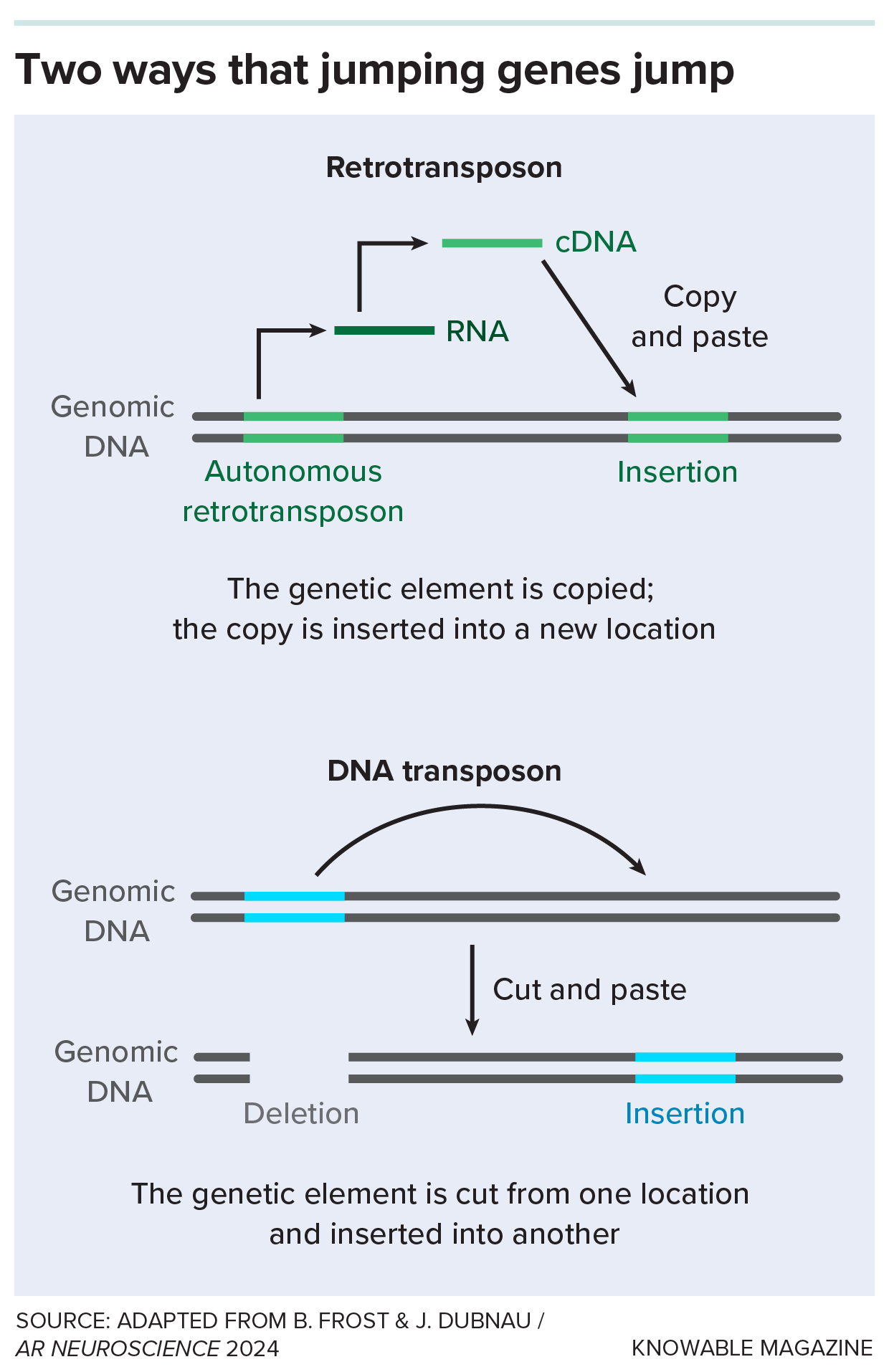
A retrotransposon is a kind of “jumping gene.” These pieces of DNA can (or once could) move around in the genome by either copying or removing themselves from one spot and then pasting themselves into a new spot. Retrotransposons are copy-and-pasters.
Many retrotransposons are old companions: Some predate the evolution of Homo sapiens or even the split between plants and animals, Dubnau says. Their predecessors may have alternated between riding along stitched into a host chromosome and existing outside of it, he suggests.
Some retrotransposons, after all that time, retain their ability to hop around human DNA. To do so, they copy themselves with the enzyme reverse transcriptase, which is also used by some viruses like HIV to copy RNA sequences into DNA. Once they’re copied, the remnant viruses can pop into new locations on chromosomes.
If it’s terrifying to think of a genome littered with retroviral genes, some capable of bouncing around the genome, don’t fret, says Douville, now at the University of Manitoba in Winnipeg. Remarkably, some retrotransposons have taken on helpful jobs, assisting the body with tasks like maintaining stem cells and development of the embryo and nervous system.
And many retrotransposons are dormant or broken, and the cell has means to keep them (mostly) quiet. One technique is to stash them in DNA regions that are wound up so tight that the molecular machines needed to copy genes can’t get near them.
In essence, the cell shoves them into a closet and slams the door shut.
But evidence is building that as people age, that closet door can creak open, letting retrotransposons spill out. Exactly what they do then isn’t certain. Some scientists think it’s not so much that they are jumping around and mutating DNA, but that their viralesque RNAs and proteins can screw up normal cellular activities.
“I think what’s actually driving toxicity when transposons are activated is they’re making all these factors that look like a virus to the cell,” says Bess Frost, a neurobiologist at Brown University in Providence, Rhode Island. The cell reacts, quite reasonably, with defensive inflammation, which is commonly associated with neurodegeneration.
Retrotransposons also seem to team up with rogue proteins classically linked to neurodegeneration, damaging or killing nerve cells, and perhaps even setting off the disease in the first place.
Making the ALS connection
Scientists long suspected a link between viruses and ALS, which causes degeneration of the motor neurons that control movement. But the connection, when it was finally found, wasn’t quite what anyone predicted.
In the early 2000s, scientists reported that some people with ALS had the viral enzyme reverse transcriptase in their blood and, more rarely, spinal fluid. Some had as much reverse transcriptase as a person with an HIV infection.
But at the time, says Dubnau, “Nobody could find a virus.”
Finally, Douville and colleagues discovered evidence for one of those leftover viruses, a kind of retrotransposon called HERV-K, in the brains of some people who had died of ALS. From there, scientists began to build a case linking jumping genes to ALS in people, lab animals and cells in dishes. A team reported in 2017 that numerous jumping genes had been activated in the brains of certain people with ALS.
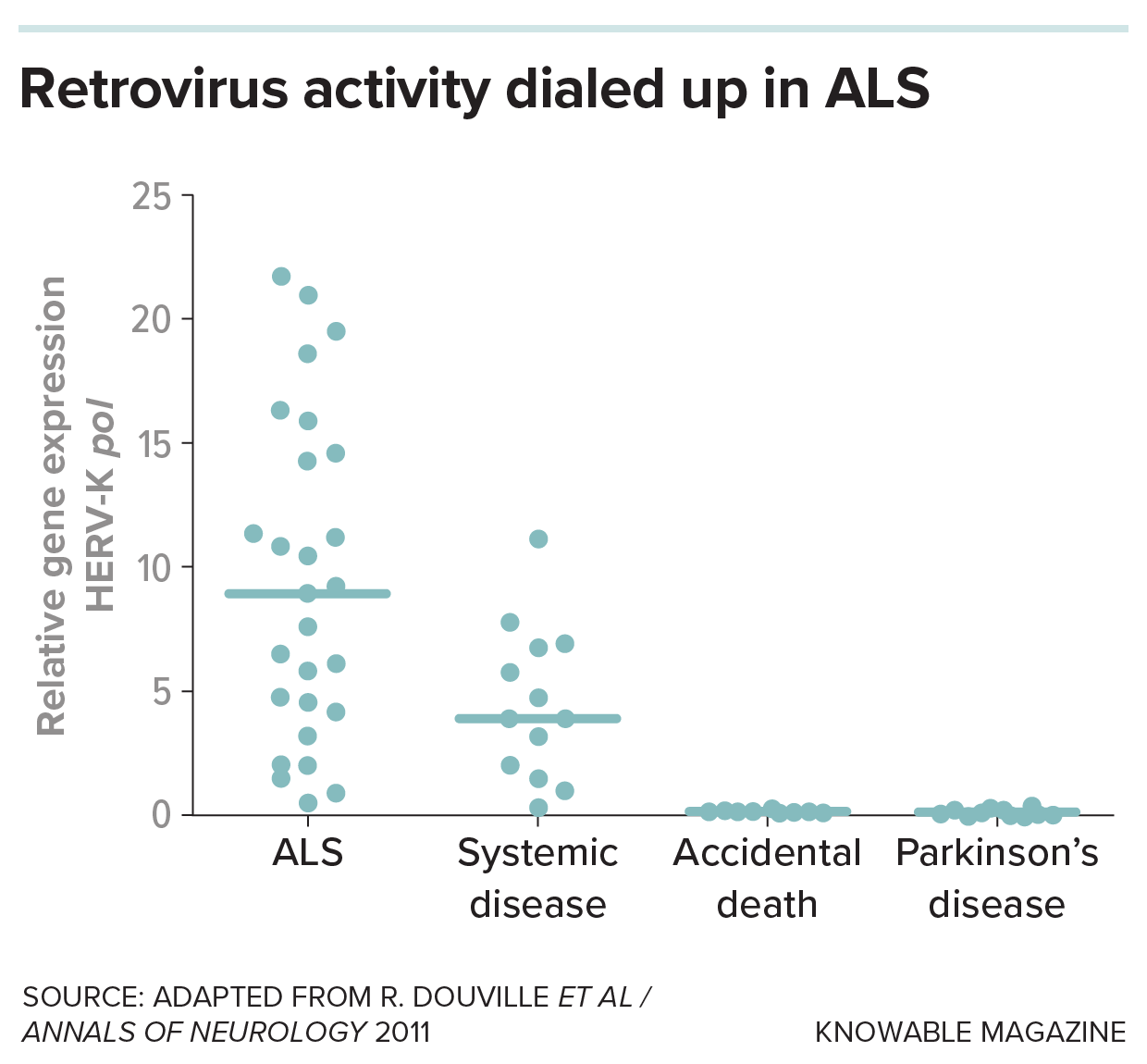 Douville’s colleagues also documented damage inflicted by HERV-K: When they put a gene from the retrotransposon into mice, the animals’ nerve cell projections shriveled and they exhibited ALS-like symptoms.
Douville’s colleagues also documented damage inflicted by HERV-K: When they put a gene from the retrotransposon into mice, the animals’ nerve cell projections shriveled and they exhibited ALS-like symptoms.
As the scientists zeroed in on what might be waking up HERV-K, a familiar protein turned up. Called TDP-43, it had already been linked to ALS. But even before that, it was found to be involved in cells’ responses to the retrovirus HIV.
Scientists discovered in the 1990s that TDP-43 works in the cell’s nucleus, where it hinders activation of HIV genes. It also regulates human genes there. But in the neurons of people with ALS or a related condition, frontotemporal dementia (FTD), TDP-43 departs the nucleus and goes on to form abnormal clumps in the cytoplasm. The globs have been associated with a number of neurodegenerative conditions and can spread from cell to cell. And when TDP-43 vacates the nucleus, it also creates a gap in gene regulation, throwing off the activity levels of many genes.
TDP-43 gone bad is sufficient to cause neurodegeneration, but studies indicate its desertion of its nuclear role can also wake up retrotransposons. When TDP-43 leaves the nucleus, tightly coiled DNA next to certain retrotransposons starts to loosen up and unravel, a study of cells from the brains of people who died of ALS or FTD revealed. And researchers saw that in cultured cells, this loss of TDP-43 freed certain retrotransposons from their restraints. The closet door was now ajar, in other words, allowing the retrotransposons to jump out and around.
Meanwhile, Dubnau and collaborators, were looking at data on TDP-43 and the genes it controls in rats, mice and people. They found that TDP-43 can naturally stick to the RNAs of a variety of jumping genes, suggesting a way that normal TPD-43 might continue to corral them, even if they’ve managed to get copied into RNA. That interaction was altered in people with FTD and in rodents with abnormally high or low amounts of TDP-43 — very much as if TDP-43 was unable to control the jumping genes anymore.
The Dubnau group also turned to fruit flies. Both old age and the human TDP-43 gene caused retrotransposons in the fly brain to sneak out of the chromosomal closet, inducing brain cells to kill their neighbors and prompting neurodegeneration, the group reported in a series of papers from 2013 to 2023. Moreover, activation of certain retrotransposons also caused TDP-43 to clump together outside of the nucleus, creating a vicious cycle whereby TDP-43 and the retrotransposons reinforce each other’s abnormal behaviors. Past a certain point, says Dubnau, “It just takes off.”
Based on the sum of all these findings, Dubnau suggests a possible way that ALS could develop: Normally, TDP-43 in the nucleus helps to repress retrotransposons. But if aging or some other disturbance causes TDP-43 to decamp, those once-silenced retrotransposons spring to life, producing virus-like RNAs and proteins. While the retrotransposons might induce disease on their own, by jumping into new DNA locations or spurring inflammation, they also act on TDP-43. They force more TDP-43 to leave the nucleus and clump in the cytoplasm, causing further neurodegeneration that spreads to neighboring cells.
This isn’t the cause of all kinds of ALS, which is a complex disorder with many possible triggers. But in a 2019 study of postmortem brain samples, Dubnau and colleagues found that about one in five people with ALS had high levels of retrotransposon activation and TDP-43 dysfunction.
The Wire is now on WhatsApp. Follow our channel for sharp analysis and opinions on the latest developments.



